新生儿遗传代谢疾病筛查 (四项)
目前,我省新生儿疾病筛查病种主要包括苯丙酮尿症 (PKU) 、先天性甲状腺功能减低症 (CH) 和G6PD酶缺乏症、17羟孕酮(CAH)等4种常见新生儿遗传代谢性疾病。

高苯丙氨酸血症
高苯丙氨酸血症 (HPA) 的定义:
口血苯丙氨酸 ( Phe ) 持续 >120umol/L ( >2mg/dl)
口血苯丙氨酸/酪氨酸 (Tyr) 比值 (Phe/ Tyr ) >2.0

常染色体隐性遗传性疾病、氨基酸代谢障碍性疾病
苯丙氨酸羟化酶 (PAH) 基因突变

苯丙氨酸 (Phe) 及其产物蓄积发病

智力落后、皮肤毛发色素浅淡、鼠尿味
Phe代谢途径
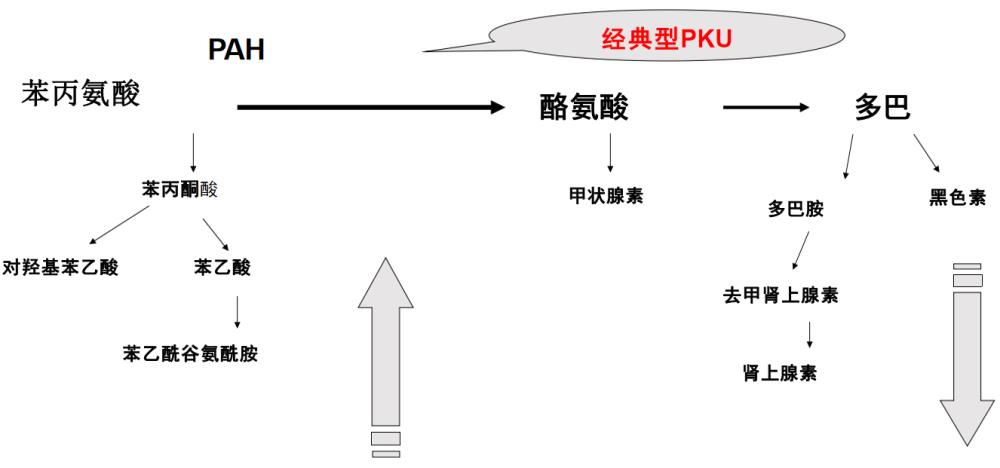
Phe代谢途径
临床表现
1 、出生时正常,部分可出现喂养困难、呕吐、易激惹等非特异性症状。未治疗的患 儿3-4 个月开始出现发育落后,头发变黄,皮肤白,全身和尿液有特殊鼠臭味,常有湿疹。
2 、典型病例:程度不同的智能低下,头发皮肤颜色变浅,尿液、汗液中散发鼠臭 味,伴有精神行为异常。
3 、除影响智能发育外,还可出现行为、性格异常,如:忧郁、多动、 自卑、孤独自闭等。


60%年长儿有严重智能障碍 (IQ低于50)
2/3患儿轻微神经系统受损体征,如肌张力增高、腱反射亢进、小头畸形等。
¼患儿有癫痫发作,可表现为婴儿痉挛性发作、点头样发作或其他形式。
80%患儿有脑电图异常,以痫性放电为主。
实验室检查
1 、新生儿疾病筛查:哺乳3-7天,采足跟血,滴在滤纸片上干燥送检

2 、Phe浓度测定 (串联质谱谱法)
切割值:<120umol/L(2mg/dl)
经典PKU: >1200umol/L(20mg/dl)
中度PKU: 360~1200umol/L(6-20mg/dl) 轻度HPA: 120~360umol/L(2-6mg/dl)
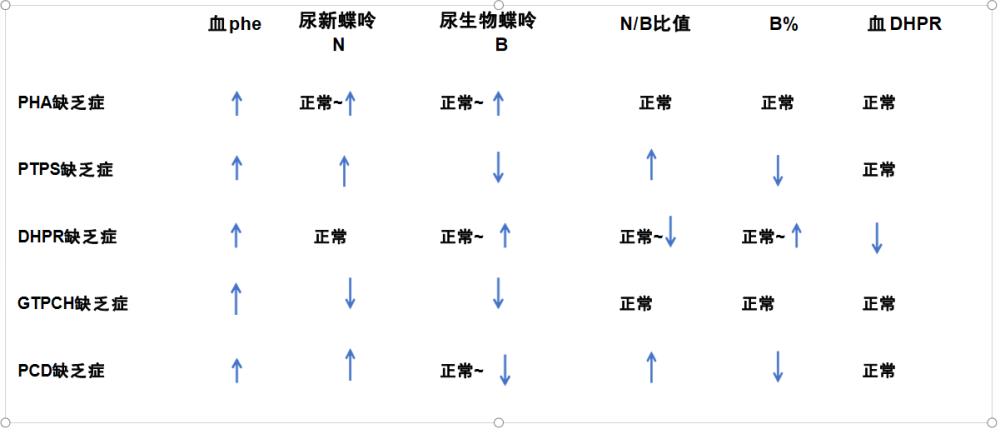
3、尿蝶呤谱: (高效液相色谱法)
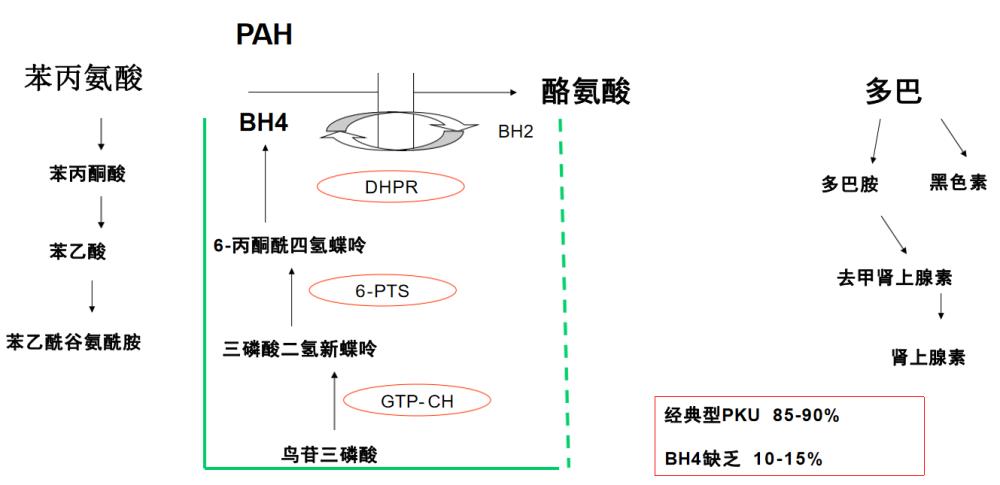
不同病因导致的HPA生化特点
血苯丙氨酸:1mg/dl=60umol/L
4 、酶学诊断:
PAH仅存于肝细胞内,不能用于临床诊断;DHPR 、GTP-CH 、6-PTS可 采用外周血红白细胞或皮肤成纤维细胞测定。
DHPR缺乏症者活性极低,多为正常者<10%。
5 、基因检测:
(1) 、PAH基因:位于染色体12q22-24.1 ,全长约90Kb,含13个外显子,编码451个氨基酸。至今国际上已报道近800种PHA 基因 突变类型。
(2) 、BH4相关基因:
PTPS酶基因位于11q22.3-q23.3 ,包括6个外显子,东亚地区已发现43种, 中国 PTS基因热点突变为c.155A>c.259C>T 、c.286G>A和· c.IVSI-291A>G。
DHPR基因位于4p15.3 ,含7个外显子,已报道35种基因突变类型。
6 、BH4负荷试验:
意义:BH4缺乏症的辅助诊、BH4反应型PKU/HPA的判断
方法:口服BH4片20mg/kg,服药前、服药后2、4、6、8、24小时分别取血作Phe测定。
新生儿:基础phe>400umol/L,可在喂奶前30分钟口服BH4片2天或更长时间:对于尿碟呤及DHPR活性正常患儿,此试验有助于鉴别BH4反应型
判断标准:5次血Phe浓度下降30%为有反应,见于BH4缺乏或部分BH4反应型PKU/HPA。
7 、脑电图:约80%的PKU患儿脑电图异常,可表现为高峰戒律紊乱、灶性 棘波等。随着治疗后Phe浓度下降,异常脑电图改变会逐渐好转。
8 、CT和MRI检查:可发现不同程度脑发育不良,表现为脑皮质萎缩、 脑白质脱髓鞘病变,在MRI的T1加权图像上显示脑室三角区周围组织条 形或斑片状高信号区。
9 、智力测定:评估智能发展程度。
诊断
1 、苯丙氨酸羟化酶 (PAH) 缺乏(85-90%) 经典PKU: >1200umol/L(20mg/dl)
中度PKU: >360~1200umol/L(6-20mg/dl)
轻度HPA: 120~360umol/L(2-6mg/dl)
BH4反应型
BH4无反应型
2 、PAH辅酶- 四氢生物碟呤 (BH4) 缺乏 (10-15%)
PTPS缺乏 (59%)
GTP-CH缺乏 (4%)
DHPR缺乏 (32%)
治 疗
1 、一旦确诊,应立即治疗。年龄越小,预后越好。
2 、血Phe应控制在一定范围,以满足生长需要。
3 、因每个患儿对Phe的耐受量不同,个体化治疗。 治疗至少持续到青春期,提倡终身治疗。
4 、家长的积极合作是成功的关键因素之一。
5 、对于成年女性PKU患者如果不控制饮食就怀孕,其后代虽然不是PKU , 仍可出现智能落后、小头畸形、先天性心脏病、 自然流产、出生低体重儿等, 称为母源性PKU综合征。
为避免此类事件发生,应告知女性PKU患者怀孕之前期半年起严格控制血Phe浓度在 120-360umol/L (2-6mg/dl) ,直至分娩,以避免高苯丙氨酸对胎儿的影响。
6 、PKU高危家庭产前诊断:通过直接查找PAH基因突变位点,或 者结合遗传多态性分析,成功地对高危家系实施产前诊断,取得 良好的社会效益。
产前诊断之前必须事先采集PKU患儿及其父母静脉血做家系突变分析,明确患儿突 变类型。于孕9- 12周取绒毛或孕17- 18周取羊水细胞进行DNA分析。由于不是直接检测基 因突变,因此必须注意临床诊断的准确性,不能将非PHA基因突变的病例来进行连锁分 析诊断。另外还必须严防样品污染,尤其是母源性有核细胞污染。
先天性甲状腺功能减低症
概 述
先天性甲状腺功能减低症:
Congenital Hypothyroid ism CH
是因先天性甲状腺发育异常或代谢障碍引起血 液循环中甲状腺激素减少的一种较常见的内分泌疾病。
早期症状不典型,常造成诊治延误。新筛避免 了严重神经损害及体格发育落后。
发病率:1/3000-5000 男/女比例1:2

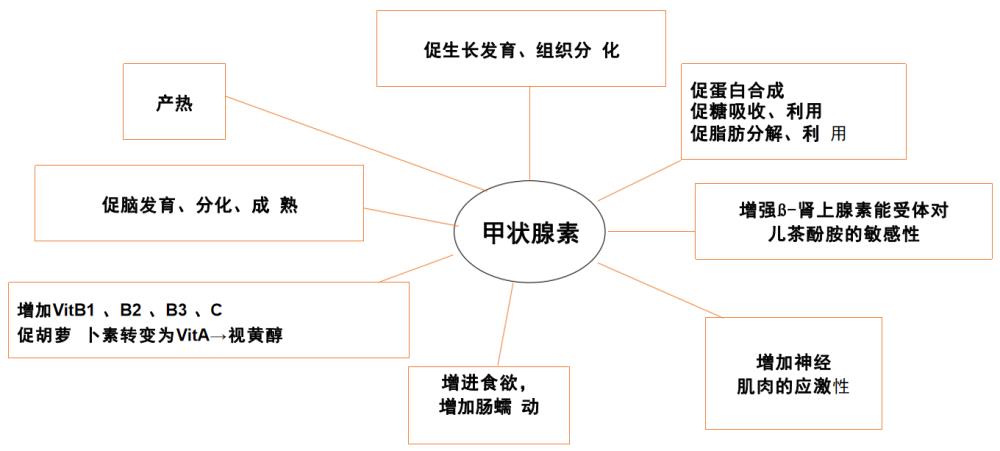
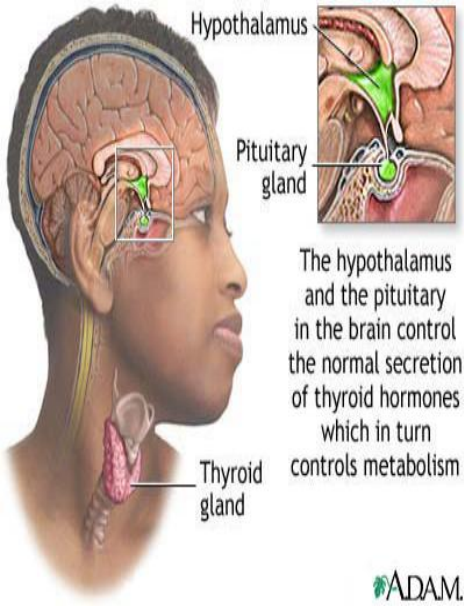
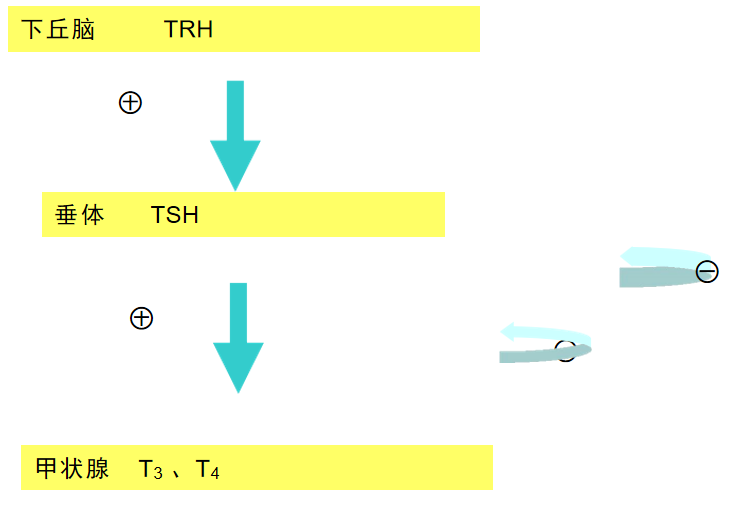
甲状腺素的调节机制
病因与分类
按病因:
散发性先天性甲减:先天性甲状腺发育不良、异位、甲状腺素合成障碍地方性先天性甲减:碘缺乏
按病变部位:
原发性先天性甲减:甲状腺 TSH,FT4
继发性先天性甲减:下丘脑和垂体 TSH正常或FT4
按疾病转归:
持续性甲减:终身替代治疗
暂时性甲减:甲状腺功能恢复正常
高TSH血症:TSH正常、持续或升高发展为甲减
甲状腺不发育、发育不全或异位:主要原因90%
甲状腺素合成 途径酶缺乏:第二位原因 常染色体隐性遗传
TRH 、TSH缺乏:特发性垂体功能低下或下丘脑、垂体发育缺陷
甲状腺或靶器官反应低下:罕见
临床表现
|
新生儿期:
过期产,体重大; 前囟大,排便迟; 腹泻便秘和脐疝; 黄疸延,反应低; 体温低,哭声低; 四肢冷,易误诊。 |
婴幼儿期:症状出生半年后出现
①特殊面容和体态:
头大、颈短、肤粗糙;
面黄、发稀、无光泽;
眼面水肿、鼻低平;
唇厚、舌大、伸口外;
身材矮小,四肢短;
腹部膨隆,伴脐疝。
②智能低下,运动发育延迟;
③生理功能低下:
少吃、少哭、少动;
体温低、心音低、肌张力低

新生儿筛查
早期发现、早期诊断的必要手段
方法:足月新生儿出生72h后,7d之内,并充分哺乳,足跟采血,滴于 专用滤纸片上测定滤纸片TSH值。
优点:能检出原发性甲减和高TSH血症
缺点:无法检出中枢性甲减、TSH延迟升高
5%的筛查漏诊率 (技术和个体差异)
危重新生儿或接受过输血治疗的新生儿可能出现假阴性
低或极低体重儿由于下丘脑-垂体- 甲状腺抽反馈建立延迟,可能出现TSH延 迟升高,可在生后2-4周或体重超过2500g时重新采血复测
|
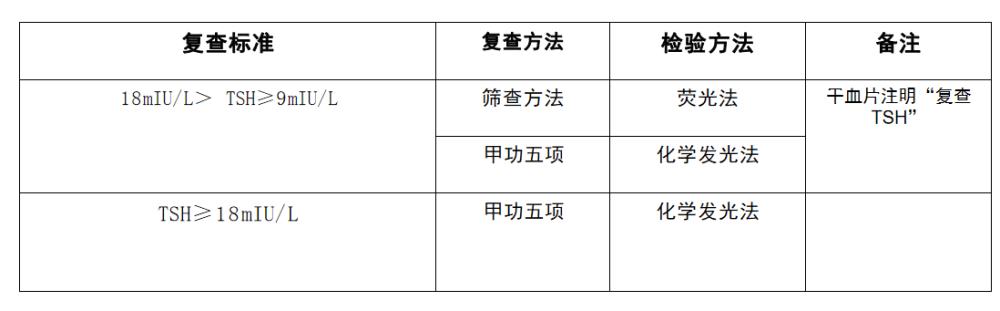
可疑阳性结果 足跟血 TSH: ≥9mIU/L |

确诊性检查
主要以TSH 、FT4为实验室确诊指标,而T4、T3为辅助确诊指标。
TSH增高、FT4降低:先天性甲状腺功能减低症 ØTSH增高、FT4正常:高TSH血症
TSH正常或降低,FT4降低:继发性或中枢性甲减
其他辅助检查
骨龄:胫骨骨化中心
甲状腺同位核素检查:125I或99mTc扫描 可检测移位、发育不良或缺如
甲状腺B超:检查甲状腺是否缺失、大小形态及位置
甲状腺球蛋白 (Tg) :反映甲状腺组织存在与否及其活性
Tg↑+摄碘不足:TSH受体突变、碘转运障碍或母源性TRB-Ab
Tg ↓:甲状腺发育不良
抗甲状腺抗体检测:孕母患自身免疫性甲状腺疾病,母子均测TGAb 、TRBAb
基因学检查
仅在有家族史或其他检查物提示为某种缺陷的甲低时进行
治 疗

治 疗----停药
甲状腺缺如、移位、发育不良者---终身服药
其他患儿可在正规治疗2-3年后尝试停药1月观察
FT4低、TSH升高—永久性甲减,恢复治疗; FT4及TSH正常— 暂时性甲减,随诊
服药剂量大者,为防止停药引起的严重损伤,可先减半量1 月后复查 1月后TSH超过20mIU/L ,--永久性甲减,恢复治疗; 1月后TSH没有升高,FT4及TSH正常— 暂时性甲减,停药观察
随 访
定期监测甲功
定期进行听力评估:CH患儿常存在听力障碍
监测心血管系统: CH患儿常合并先天性心脏病 (威廉姆斯综合征)
定期监测体格及智力发育
红细胞葡萄糖-6-磷酸脱氢酶 (G-6-PD) 缺乏症
概 述
G-6-PD缺乏症是人类最常见的单基因遗传病之一,属于X伴性不完全显性遗传性疾病, 因基因突变导致红细胞G-6-PD酶活性降低而发病。
据统计,全球共2.2亿男性及1.3亿女性患者,占世界人口4.9%。
本病分布与疟疾的选择性有关,在我国,本病呈现“南高北低”趋势, 海南、广西、广东、云南、贵州患病率高,4-20%。
发 病 机 制
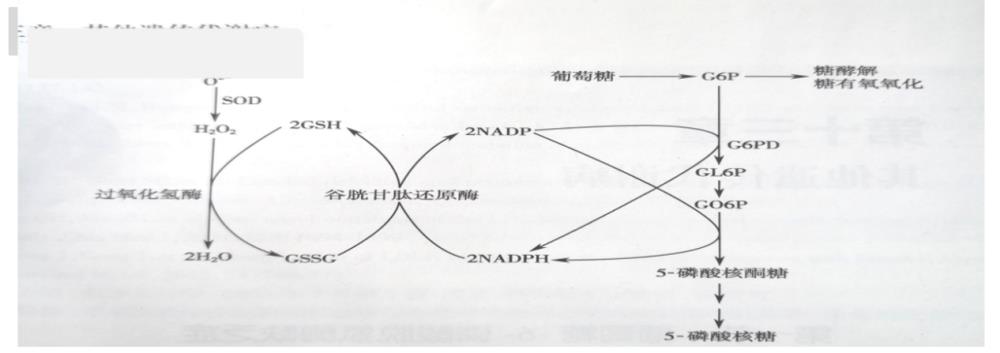
磷酸戊糖旁路途经
6-磷酸葡萄糖在G6PD酶的催化下转化为6-磷酸葡萄糖酸和NADPH (二氢烟酰胺腺嘌呤二核苷酸) ,NADPH催化GSSG (氧化型谷胱甘肽) 转化为GSH (还原型谷胱甘肽) ,GSH使得过氧化氢 (H2O2 ) 脱氧,避免其对细胞产生氧化作用。 GSH (还原型谷胱甘肽) 是细胞抗氧化保护因子,清除细胞内过氧化氢,保护细胞免受氧化损害。
发 病 机 制
蚕豆嘧啶、共蚕豆嘧啶核苷

产生过氧化氢,诱发体内氧化反应

红细胞破坏、溶血
临床表现
G-6-PD缺乏症患者酶活性等级及临床表现

本病一般无症状
急性发作性溶血 (常见)
诱因下发生严重急性溶血综合征:血红蛋白尿、贫血、黄疸、严重肾衰竭。
溶血程度与诱因、摄入物质数量、患者酶活性缺乏程度有关,药物诱发的溶血通常具有自限性。
慢性非球形红细胞性溶血性贫血 (少见)
轻- 中度贫血,可因感染、服药而加重,常伴有肝脾肿大、黄疸
新生儿严重高胆红素血症及核黄疸
G-6-PD缺乏者发生核黄疸的风险大于非G-6-PD缺乏者
实验室检查
新生儿筛查:
荧光法筛查;直接测酶活性测定 (高铁血红蛋白还原实验、荧光斑点法、硝基四氮唑蓝法、定量比值法)
常规检查:
正细胞正色素性贫血,网织红细胞升高、突变红细胞增多;骨髓象增生活跃;尿潜血阳性,可见蛋白、红 细胞、管型、尿胆原、尿胆素
基因诊断:
男性半合子及女性纯合子酶活性程度约为正常人的5%左右,普通酶活性检测可被检出;
女性杂合子酶活性程度可正常或轻度降低,但有可能遗传给子代,基因检测可以提供重要信息。
治疗及预防
本病无特殊治疗方法,新筛是早期诊断、预防的重点。
急性溶血时按急性溶血性贫血的处理原则治疗。
避免接触氧化性药物和食物。
本病属于遗传性可预防性疾病,有一定诱因才发病,一般不对胎儿进行产前诊断。

